Hi all,
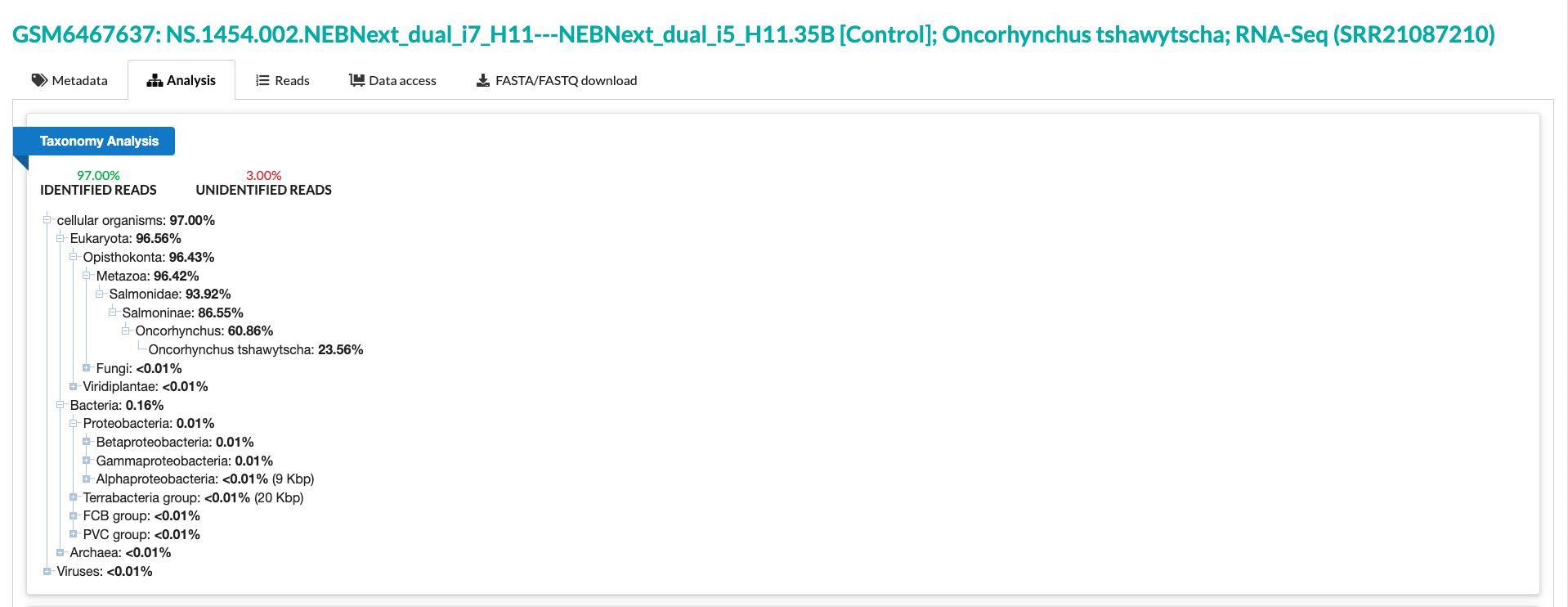
I am running humann3 for some of my samples and I keep getting empty reads. The samples are from the fish gut and I did RNA sequencing to see how antibiotics and probiotics can change host gene expression. But now I found out that by using Humann3 or Kraken I can get the microbial community as well (while expecting to lose a lot of host reads). So I used Knead data to get rid of the host genes and now while I am using the output from Kneaddata, the humann3 output is empty. Here is a detailed output of my command. I also uploaded my sequences on NCBI SRA and from the NCBI database I can see at least one million reads should be for the microorganism (please see the screenshot or this link (https://trace.ncbi.nlm.nih.gov/Traces/index.html?view=run_browser&acc=SRR21087210&display=analysis). And also I can get results using Kraken.
05/02/2023 01:11:26 PM - humann.humann - INFO: Running humann v3.6
05/02/2023 01:11:26 PM - humann.humann - INFO: Output files will be written to: /home/jsadeghi/scratch/Javad/Ch4/2_Humann
05/02/2023 01:11:26 PM - humann.humann - INFO: Writing temp files to directory: /home/jsadeghi/scratch/Javad/Ch4/2_Humann/35B_cat_humann_temp
05/02/2023 01:11:26 PM - humann.utilities - INFO: File ( /home/jsadeghi/scratch/Javad/Ch4/1_Knead_data_cleaned/35B_cat ) is of format: fastq
05/02/2023 01:11:26 PM - humann.utilities - DEBUG: Check software, metaphlan, for required version, 3.0
05/02/2023 01:12:09 PM - humann.utilities - INFO: Using metaphlan version 3.0
05/02/2023 01:12:09 PM - humann.utilities - DEBUG: Check software, bowtie2, for required version, 2.2
05/02/2023 01:12:11 PM - humann.utilities - INFO: Using bowtie2 version 2.5
05/02/2023 01:12:11 PM - humann.humann - INFO: Search mode set to uniref90 because a uniref90 translated search database is selected
05/02/2023 01:12:11 PM - humann.utilities - DEBUG: Check software, diamond, for required version, 2.0.15
05/02/2023 01:12:11 PM - humann.utilities - INFO: Using diamond version 2.0.15
05/02/2023 01:12:11 PM - humann.config - INFO:
Run config settings:
DATABASE SETTINGS
nucleotide database folder = /scratch/st-spakpour-1/envs/humann3/installed_databases/chocophlan
protein database folder = /scratch/st-spakpour-1/envs/humann3/installed_databases/uniref
pathways database file 1 = /scratch/st-spakpour-1/envs/humann3/lib/python3.8/site-packages/humann/data/pathways/metacyc_reactions_level4ec_only.uniref.bz2
pathways database file 2 = /scratch/st-spakpour-1/envs/humann3/lib/python3.8/site-packages/humann/data/pathways/metacyc_pathways_structured_filtered_v24
utility mapping database folder = /scratch/st-spakpour-1/envs/humann3/installed_databases/utility_mapping
RUN MODES
resume = False
verbose = False
bypass prescreen = False
bypass nucleotide index = False
bypass nucleotide search = False
bypass translated search = False
translated search = diamond
threads = 1
SEARCH MODE
search mode = uniref90
nucleotide identity threshold = 0.0
translated identity threshold = 80.0
ALIGNMENT SETTINGS
bowtie2 options = --very-sensitive
diamond options = --top 1 --outfmt 6
evalue threshold = 1.0
prescreen threshold = 0.01
translated subject coverage threshold = 50.0
translated query coverage threshold = 90.0
nucleotide subject coverage threshold = 50.0
nucleotide query coverage threshold = 90.0
PATHWAYS SETTINGS
minpath = on
xipe = off
gap fill = on
INPUT AND OUTPUT FORMATS
input file format = fastq
output file format = tsv
output max decimals = 10
remove stratified output = False
remove column description output = False
log level = DEBUG
05/02/2023 01:12:11 PM - humann.store - DEBUG: Initialize Alignments class instance to minimize memory use
05/02/2023 01:12:11 PM - humann.store - DEBUG: Initialize Reads class instance to minimize memory use
05/02/2023 01:12:33 PM - humann.humann - INFO: Load pathways database part 1: /scratch/st-spakpour-1/envs/humann3/lib/python3.8/site-packages/humann/data/pathways/metacyc_reactions_level4ec_only.uniref.bz2
05/02/2023 01:12:33 PM - humann.humann - INFO: Load pathways database part 2: /scratch/st-spakpour-1/envs/humann3/lib/python3.8/site-packages/humann/data/pathways/metacyc_pathways_structured_filtered_v24
05/02/2023 01:12:33 PM - humann.search.prescreen - INFO: Running metaphlan …
05/02/2023 01:12:33 PM - humann.utilities - DEBUG: Using software: /scratch/st-spakpour-1/envs/humann3/bin/metaphlan
05/02/2023 01:12:33 PM - humann.utilities - INFO: Execute command: /scratch/st-spakpour-1/envs/humann3/bin/metaphlan /home/jsadeghi/scratch/Javad/Ch4/1_Knead_data_cleaned/35B_cat -t rel_ab -o /home/jsadeghi/scratch/Javad/Ch4/2_Humann/35B_cat_humann_temp/35B_cat_metaphlan_bugs_list.tsv --input_type fastq --bowtie2out /home/jsadeghi/scratch/Javad/Ch4/2_Humann/35B_cat_humann_temp/35B_cat_metaphlan_bowtie2.txt
05/02/2023 01:16:16 PM - humann.utilities - DEBUG: b’’
05/02/2023 01:16:16 PM - humann.humann - INFO: TIMESTAMP: Completed prescreen : 223 seconds
05/02/2023 01:16:16 PM - humann.search.prescreen - INFO: Total species selected from prescreen: 0
05/02/2023 01:16:17 PM - humann.search.prescreen - DEBUG:
No species were selected from the prescreen.
Because of this the custom ChocoPhlAn database is empty.
This will result in zero species-specific gene families and pathways.
05/02/2023 01:16:17 PM - humann.humann - INFO: TIMESTAMP: Completed custom database creation : 0 seconds
05/02/2023 01:16:17 PM - humann.humann - DEBUG: Custom database is empty
05/02/2023 01:16:17 PM - humann.store - DEBUG: Initialize Reads class instance to minimize memory use
05/02/2023 01:18:01 PM - humann.utilities - DEBUG: Remove file: /home/jsadeghi/scratch/Javad/Ch4/2_Humann/35B_cat_humann_temp/tmp6xxu3ry_/tmp4hjr3d5m
05/02/2023 01:18:01 PM - humann.search.translated - DEBUG: Convert unaligned reads fastq file to fasta
05/02/2023 01:19:19 PM - humann.search.translated - INFO: Running diamond …
05/02/2023 01:19:19 PM - humann.search.translated - INFO: Aligning to reference database: uniref90_201901b_full.dmnd
05/02/2023 01:19:19 PM - humann.utilities - DEBUG: Remove file: /home/jsadeghi/scratch/Javad/Ch4/2_Humann/35B_cat_humann_temp/tmp6xxu3ry_/diamond_m8_hy991q3u
05/02/2023 01:19:19 PM - humann.utilities - DEBUG: Using software: /scratch/st-spakpour-1/envs/humann3/bin/diamond
05/02/2023 01:19:19 PM - humann.utilities - INFO: Execute command: /scratch/st-spakpour-1/envs/humann3/bin/diamond blastx --query /home/jsadeghi/scratch/Javad/Ch4/2_Humann/35B_cat_humann_temp/tmp6xxu3ry_/tmpwzdj83dr --evalue 1.0 --threads 1 --top 1 --outfmt 6 --db /scratch/st-spakpour-1/envs/humann3/installed_databases/uniref/uniref90_201901b_full --out /home/jsadeghi/scratch/Javad/Ch4/2_Humann/35B_cat_humann_temp/tmp6xxu3ry_/diamond_m8_hy991q3u --tmpdir /home/jsadeghi/scratch/Javad/Ch4/2_Humann/35B_cat_humann_temp/tmp6xxu3ry_